

20 de fevereiro de 2014
17 de janeiro de 2014
Doenças Císticas do Rim
As doenças císticas do rim têm importância significativa por sua relativa frequência e dificuldade de diagnóstico tanto para clínicos, como para patologistas e radiologistas, podendo ser confundidas com tumores malignos. Algumas de suas formas, como a Doença Policística do Adulto, são responsáveis por quadros de Insuficiência Renal Crônica, como veremos adiante. A maioria das doenças císticas apresenta uma alteração genética envolvida, podendo ter caráter hereditário ou não, mas outras não apresentam alterações genéticas características.
As doenças císticas do rim são:
¨Displasia renal cística;
¨Doença renal policística;
¨Doença cística medular;
¨Doença cística adquirida;
¨Cistos renais localizados;
¨Cistos renais em síndromes de malformações hereditárias;
¨Doença glomerulocística;
¨Cistos
renais extraparenquimatosos.
As principais doenças (em vermelho), serão discutidas a seguir.
DISPLASIA RENAL CÍSTICA
As principais características são:
¨Anormalidade na diferenciação metanéfrica;
¨Persistência de estruturas anormais - cartilagem, mesênquima não diferenciado e túbulos coletores
imaturos -
e organização lobar anormal;
¨Pode estar associada à obstrução ureteropélvica, agenesia uretral ou
atresia e outras anomalias
do sistema urinário inferior;
¨Pode ser unilateral ou bilateral (na unilateral, a
função do rim oposto é normal; na bilateral, pode ocorrer insuficiência renal).
Macroscopicamente o rim encontra-se aumentado, extremamente irregular e multicístico.
 |
| Figura 1: Macroscopia de um rim evidenciando os múltiplos cistos e o relevo irregular. Fonte: http://library.med.utah.edu/WebPath/RENAHTML/RENAL043.html |
 |
| Figura 2: Microscopia de um cisto. Fonte: http://www.fcm.unicamp.br/deptos/anatomia/casosdeuro/caso321/index1.html |
 |
| Figura 3: Microscopia evidenciando os ductos imaturos. Fonte: http://www.fcm.unicamp.br/deptos/anatomia/casosdeuro/caso321/index1.html |
 |
| Figura 4: Microscopia evidenciando as ilhotas de mesênquima com presença de cartilagem. Fonte: http://www.fcm.unicamp.br/deptos/anatomia/casosdeuro/caso321/index1.html |
Trata-se de uma doença genética com alta penetrância que afeta cerca de 400 a cada 1000 nascidos vivos. É responsável por cerca de 5 - 10% dos casos de Insuficiência Renal Crônica que necessitam de diálise ou transplante. Nela, múltiplos cistos se expandem em ambos os rins até destruir o parênquima. Casos unilaterais são mais brandos envolvendo parte dos néfrons e a função renal é mantida até a quarta ou quinta década de vida.
Está relaciona com mutações nos genes PKD1 (policistina 1) e PKD2 (policistina 2):
¨PKD1 – 85% dos casos; doença mais severa; doença renal
terminal em uma média de 53 anos de idade;
¨PKD2 – mais leve; doença renal terminal em uma média de
69 anos de idade.
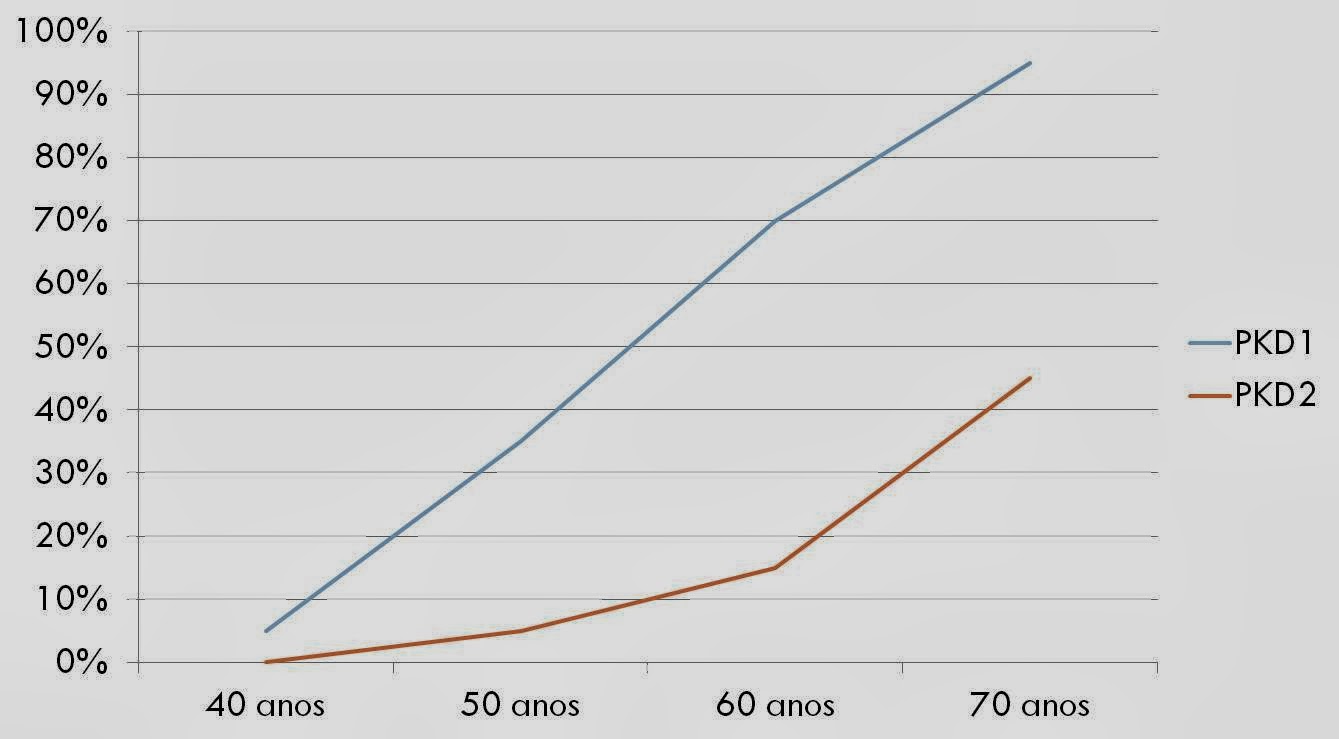 |
| Gráfico 1: Mostra a chance de desenvolvimento de insuficiência renal de acordo com a idade e com o gene mutado. |
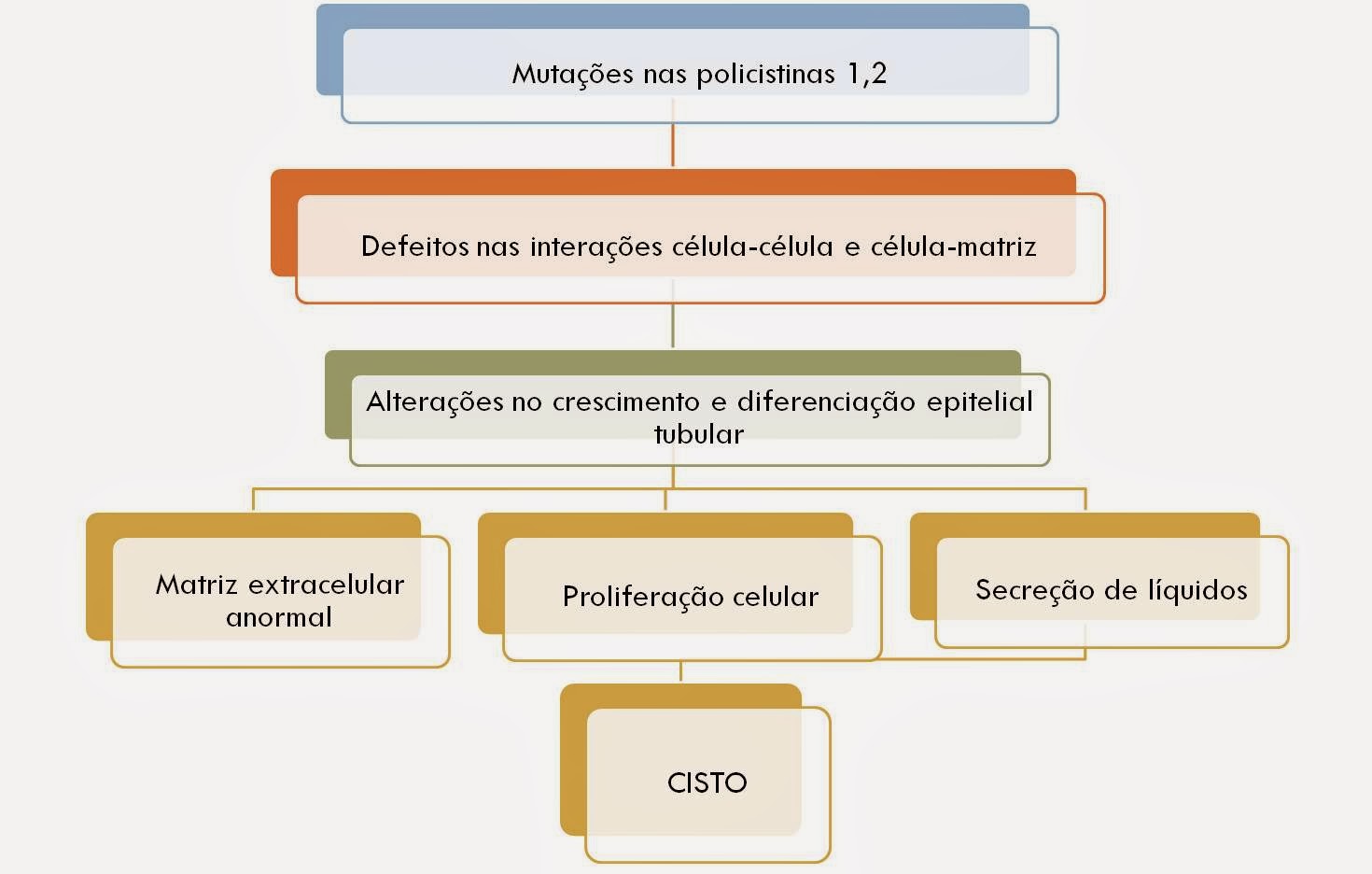 |
| Figura 5: Esquema mostrando a sequência de eventos que levam à formação dos cistos. |
 |
| Figura 6: Corte de rim evidenciando os múltiplos cistos. Note a destruição do parêquima renal. Fonte: http://library.med.utah.edu/WebPath/RENAHTML/RENALIDX.html#6 |
¨Hematúria, seguida por: proteinúria, polúria e hipertensão;
¨A excreção de coágulos sanguíneos podem causar cólicas
renais;
¨Cerca de 40% dos pacientes apresentam cistos no fígado
(doença policística hepática);
¨Prolapso de válvula mitral e outras anomalias cardíacas ocorrem
em 20% dos pacientes.
DOENÇA RENAL POLICÍSTICA AUTOSSÔMICA RECESSIVA (DA CRIANÇA)
Doença de caráter genético que costuma se apresentar no período perinatal ou neonatal, podendo também ocorrer na infância ou juventude. Está relacionada com mutação no gene PKHD1 que codifica a fibrocistina. Acredita-se que a fibrocistina seja um receptor celular de superfície com papel na diferenciação dos ductos coletores e biliares.
Macroscopicamente os rins encontram-se aumentados com aparência externa lisa, mas, aos cortes, podemos notar a aparência em esponja com canais alongados e dilatados, substituindo a medula e o córtex.
 |
| Figura 7: Necrópsia de um feto com a doença. Perceba o tamanho aumentado e a aparência lisa externa dos rins. Fonte: http://library.med.utah.edu/WebPath/RENAHTML/RENALIDX.html#6 |
 |
| Figura 8: Corte evidenciando a aparência esponjosa do rim. Fonte: http://library.med.utah.edu/WebPath/RENAHTML/RENALIDX.html#6 |
Microscopicamente encontramos dilatação sacular ou cilíndrica de todos os ductos coletores. Os cistos tem revestimento de células cubóide, refletindo a origem a partir dos ductos coletores. Em quase todos os casos, o fígado apresenta fibrose portal, bem como proliferação dos ductos biliares portais.
 |
| Figura 9: Corte histológico evidenciando a dilatação cilíndrica dos ductos coletores. Fonte: http://library.med.utah.edu/WebPath/RENAHTML/RENALIDX.html#6 |
 |
| Figura 10: Corte histológico evidenciando a fibrose portal em paciente com a Doença Renal Policística Autossômica Recessiva. Fonte: http://library.med.utah.edu/WebPath/RENAHTML/RENALIDX.html#6 |
DOENÇAS CÍSTICA MEDULAR
As duas principais doenças desse grupo são o Rim em Esponja Medular e o Complexo Nefronoftise-doença Cística Renal.
RIM EM ESPONJA MEDULAR:
É uma doença de patogênese desconhecida e que acomete, principalmente, adultos. Trata-se de múltiplas dilatações císticas dos ductos coletores da medula.
 |
| Figura 11: Corte evidenciando o aspecto esponjoso da medula renal devido aos múltiplos cistos. Fonte: http://library.med.utah.edu/WebPath/RENAHTML/RENALIDX.html#6 |
Os genes NPH1, NPH2 e NPH3 são os genes responsáveis pela herança autossômica recessiva e os genes MCKD1 e MCKD2 da herança autossômica dominante. A lesão inicial se dá nos túbulos distais acarretando em ruptura da membrana basal tubular, seguida de atrofia tubular crônica e fibrose intersticial. Essa lesão túbulo intersticial é a causa da insuficiência renal resultante.
São reconhecidas 4 variantes:
-Esporádica
não familiar (20%);
-Nefronoftise juvenil familiar
(40%-50%);
-Displasia
renal-retiniana (15%);
-Doença
cística medular do adulto (15%).
Acredita-se que esse Complexo seja a causa mais comum de Doença Renal Terminal e crianças e jovens adultos. Nas crianças, apresenta-se inicialmente com poliúria e polidipsia. Na Nefronoftise juvenil, pode ocorrer anormalidades motoras oculares, retinite pigmentosa, fibrose hepática e anormalidades cerebelares. A progressão para insuficiência renal se dá em 5 - 10 anos.
Morfologicamente os rins são pequenos. Os cistos encontram-se localizados na junção corticomedular e são revestidos por epitélio pavimentoso ou cubóide e são, geralmente, cercados por células inflamatórias ou tecido fibroso. No córtex encontramos atrofia generalizada, espessamento das membranas basais dos túbulos contorcidos proximais e distais e fibrose intersticial. Alguns glomérulos podem estar hialinizados.
DOENÇA CÍSTICA ADQUIRIDA
Como o próprio nome já diz, a doença não tem caráter genético e está mais relacionada com a diálise. Os cistos medem entre 0,5 - 2 cm de diâmetro revestidos por epitélio pavimentoso, por vezes hiperplásico, contendo líquido claro com cristais de oxalato de cálcio. A doença cursa com poucos sintomas, às vezes hematúria, e a complicação mais grave seria o surgimento de um carcinoma de células renais nas paredes dos cistos (observado em 10% dos pacientes).
CISTOS SIMPLES
Cistos múltiplos ou únicos, translúcidos e preenchido por líquido claro. Hemorragias associadas podem causa distensão súbita e dor. Clinicamente a sua importância se dá na diferenciação com tumores renais. Para diferenciá-los dos tumores renais basta observar que os cistos apresentam contorno liso, são avasculares e formam sinais líquidos, ao invés de sólidos, no ultrassom.
Referências:
-Robbins & Cotran; PATOLOGIA: BASES PATOLÓGICAS DAS DOENÇAS; 8ª Edição, 2010. Ed. Elsevier;
-http://library.med.utah.edu/WebPath/RENAHTML/RENAL043.html;
-http://www.fcm.unicamp.br/deptos/anatomia/casosdeuro/caso321/index1.html;
-http://library.med.utah.edu/WebPath/RENAHTML/RENALIDX.html#6.
River de Alencar Bandeira Coêlho
Acadêmico de Medicina
Liga de Patologia
Universidade Federal do Ceará
Assinar:
Comentários (Atom)