
Epidemiologia
O adenocarcinoma gástrico constitui umas das mais freqüentes malignidades a nível mundial e revela-se a segunda causa de morte por câncer. Nesse contexto, o Brasil aparece como um dos países considerados de alta incidência, principalmente nas regiões sudeste e nordeste [NEVES FILHO et al., 2010]. A incidência dessa neoplasia varia muito, sendo particularmente alta em países como Japão, China, Chile e Portugal e quatro a seis vezes menos comum nos Estados Unidos, Canadá e Franca. Apresenta-se mais em grupos socioeconômicos mais baixos e exibe uma relação homem/mulher de aproximadamente 2/1 [KUMAR; ABBAS; FAUSTO, 2005].
Classificação
Os dois sistemas de classificações do CG mais usados de acordo com critérios microscópicos são aquele da OMS e o histopatológico de Lauren [KUMAR; ABBAS; FAUSTO, 2005] [LAUREN, 1965]. Em 1965, Lauren subdividiu o adenocarcinoma gástrico em tipo intestinal (tumores volumosos, diferenciados, de estrutura histológica glandular) e difuso (infiltrativos, indiferenciados e de células pouco coesas) [LAUREN, 1965]. O sistema proposto pela OMS vem sendo usado desde 1977, sendo bem aceito por ser simples, classificando os tumores somente com base em critérios histológicos [KUMAR; ABBAS; FAUSTO, 2005].


Carcinogênese
Os fatores envolvidos no desenvolvimento do CG variam, principalmente, de acordo com o tipo histológico segundo a classificação de Lauren e com a região anatômica de acometimento do tumor (se cárdia ou não-cárdia) (ver tabela 3) [CREW; NEUGUT, 2006], [KAMANGAR et al., 2006] . Evidências epidemiológicas apontam o subtipo difuso como de caráter mais hereditário; os fatores de risco envolvidos na patogênese deste histotipo, entretanto, permanecem desconhecidos. Os tumores de classificação intestinal estão relacionados ao desenvolvimento de gastrite atrofica com posterior metaplasia intestinal e displasia como lesões precursoras. [MELO et al., 2010], [CORREA, 1992] Fatores dietéticos, infecção por Helicobacter pylori, tabagismo e obesidade são reconhecidamente os principais fatores de risco para a carcinogênese gástrica [CREW; NEUGUT, 2006].
Biologia Molecular
Dentre as alterações genéticas descritas em CG, destaca-se a ativação dos oncogenes c-met, k-sam e c-erb. Mutações em APC tendem a ser mais freqüentes no subtipo intestinal, assim como a instabilidade de microssatélites [FERRASI et al., 2010]. Genes supressores de tumor encontram-se freqüentemente alterados, como mutação de p53 e metilação de CDKN2A, resultando em não-expressão da proteína p16 [LIMA et al., 2010], [ALVES et al., 2010]. A perda da coesão celular observada no subtipo difuso reflete redução ou perda da molécula de adesão E-caderina [GUIFORD et al., 1998].
Helicobacter pylori
H. pylori é um bacilo flagelado que coloniza a mucosa gástrica, cuja infecção desencadeia um processo inflamatório crônico, podendo levar ao desenvolvimento de lesões pré-malignas, como gastrite atrofica num estado de hipocloridria, e CG (cascata de Correa) [CORREA, 1992]. As alterações seqüenciais dependem tanto da presença de cepas bacterianas mais virulentas quanto da resposta imune organizada pelo hospedeiro. Dentre os fatores de virulência mais estudados, destaca-se a presença do gene cagA, codificador de proteína citotóxica homônima, e de variações alélicas do gene vacA, como a forma s1m1, codificador de proteína vacuolizante [ATHERTON, 1998]. Esse microorganismo foi considerado pela OMS como agente carcinogênico do tipo I, ou seja, capaz de levar a transformação maligna per se [KUMAR; ABBAS; FAUSTO, 2005].
Dieta
Alimentos preservados pelo sal e nitritos encontrados em alimentos defumados são potencialmente carcinogênicos. Consumo de comida salgada pode aumentar o risco de infecção por H. pylori e age sinergicamente para a promoção de CG. Em modelos animais, a ingestão de sal é conhecida causa de gastrite e acentua os efeitos da carcinogênese gástrica [CREW; NEUGUT, 2006]. O declínio mundial da incidência de adenocarcinoma gástrico pode ser atribuído ao advento da refrigeração, o qual diminuiu vertiginosamente o consumo de alimentos preservados no sal e aumentou o consumo de frutas e vegetais frescos [MELO et al., 2010], [CREW; NEUGUT, 2006]. Esse fato é corroborado pela diminuição do CG em países desenvolvidos, onde o uso da refrigeração e bem disseminado; no Japão, em contra-partida, as altas taxas de incidência dessa neoplasia são atribuídas ao consumo tradicional de alimentos defumados [MELO et al., 2010].
Tabela 3 – Epidemiologia do CG de acordo com sitio anatômico de acometimento

BORRMANN1

Tumor polipóide (Borrmann I) [STEWART; COOKE, 2004]
BORRMANN2

Tumor ulcerado superficial (Borrmann II) [http://www.pathology.pitt.edu/lectures/gi/stom-a/map.map?70,561]
BORRMANN3

Tumor ulcerado infiltrado (Borrmann III) [STEWART; COOKE, 2004]
BORRMANN4

Tumor infiltrativo – linite plástica (Borrmann IV) [STEWART; COOKE, 2004]
Microscopia
Tipo Intestinal de Lauren
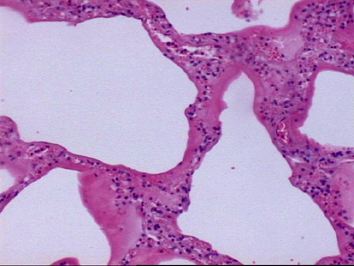
- Observar formação glandular e bem diferenciada. Note projeção papilar de algumas glândulas, com necrose nos lumens freqüentemente presente.
Lauren1

- Observar ausência de formação glandular. Há uma perda da coesão celular e um aspecto indiferenciado. Note a presença de células em anel de sinete apontada pelas setas.
Lauren2

ReferênciasTipo Intestinal de Lauren
- Observar formação glandular e bem diferenciada. Note projeção papilar de algumas glândulas, com necrose nos lumens freqüentemente presente.
Lauren1

[http://oac.med.jhmi.edu/Pathology/GI/Stomach/265A_Full.html]
Tipo Difuso de Lauren- Observar ausência de formação glandular. Há uma perda da coesão celular e um aspecto indiferenciado. Note a presença de células em anel de sinete apontada pelas setas.
Lauren2

[http://www.som.tulane.edu/classware/pathology/medical_pathology/McPath/GICD/images/Stomach/l/stomach23.jpg]
- MELO, JUS et al. NEOPLASIAS DE ESTOMAGO In: REGADAS et al. Fundamentos da Cirurgia Digestiva. Fortaleza, Edições UFC. 2010.
- CRUVEILLIER, J. Considerations generales sur les ulcerations folliculaires de l’estomac. In: Atlas d’Anatomie Pathologic. Paris: Bailliere, 1842.
- HOWSON, CP; HIYAMA, T; WYNDER, EL. The decline in gastric cancer: epidemiology of an unplanned triumph. Epidemiol Rev, v. 8, p. 1-27, 1986.
- NEVES FILHO, EH et al. MTHFR C677T polymorphism and differential methylation status in gastric cancer: an association with Helicobacter pylori infection. Virchows Archiv, v. 457, n. 6, 2010.
- KUMAR, V; ABBAS, AK; FAUSTO, N. Bases patológicas das doenças. Rio de Janeiro, Elsevier. 2005.
- LAUREN, P. The two histological main types of gastric carcinoma: diffuse and so-called intestinal-type carcinoma – na attempt at a histoclinical classification. Acta Pathol Microbiol Scan, 1965, v.64, p. 31-49.
- BORRMANN, R. Geschwuelste des magens. In: HENKE, F.; LUBARSCH, O. Handbuch der speziellen. Pathologischen anatomy und histology. Springer. Berlin 1926, p. 864-71.
- CREW, KD; NEUGUT AI. Epidemiology of gastric cancer. World J Gastroenterol, v. 12, n. 3, p. 354-62, 2006.
- KAMANGAR, F et al. Opposing risks of gastric cardia and noncardia gastric adenocarcinoma associated with Helicobacter pylori seropositivity. J Natl Cancer Inst, v. 98, n.20, p. 1445-52, 2006.
- CORREA, P. Human gastric carcinogenesis: a multistep and multifactorial process. Cancer Res. 1992, v. 52, p. 6735-40.
- FERRASI, AC et al. Helicobacter pylori and EBV in gastric carcinomas: methylation status and microsatellite instability. World J Gastroenterol. 2010 Jan 21;16(3):312-9.
- LIMA, VP et al. H pylori (CagA) and Epstein-Barr virus infection in gastric carcinomas: correlation with p53 mutation and c-Myc, Bcl-2 and Bax expression. World J Gastroenterol. 2008 Feb 14;14:884-91.
- ALVES, MK et al. CDKN2A promoter methylation is related to the tumor location and histological subtype and associated with Helicobacter pylori flaA(+) strains in gastric adenocarcinomas. APMIS. 2010 Apr;118(4):297-307.
- GUIFORD, P et al. E-cadherin germline mutations in familial gastric cancer. Nature, 1998, p. 392-402.
- ATHERTON, JC. H. pylori virulence factors. British Medical Bulletin 1998;54 (No. 1): 105-120.
- COOKE, R; STEWART, B. Colour Atlas ofAnatomical Pathology. Ed. 3rd. 2004. Londres, Elsevier.
- In web: http://www.pathology.pitt.edu/lectures/gi/stom-a/map.map?70,561
- In web: http://www.pathology.pitt.edu/lectures/gi/stom-a/map.map?70,561
- In web: http://www.som.tulane.edu/classware/pathology/medical_pathology/McPath/GICD/images/Stomach/l/stomach23.jpg
Eduardo Henrique Cunha Neves Filho
Acadêmico de Medicina
Liga de Patologia da UFC
Acadêmico de Medicina
Liga de Patologia da UFC











































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}